
- 首页 > 正文
从发病机制到诊疗指导:遗传学研究为ADPKD诊疗带来新视野
肾医线 发表时间:2025-02-17 11:57:00
编者按:ADPKD作为最常见的遗传性肾脏病,其复杂的发病机制、多变的临床表现以及挑战性的治疗策略,一直吸引着众多学者的关注。在第五届全球华人肾脏病学术大会暨香港肾科学会周年学术会议(ICCN 2024)上,加拿大多伦多大学健康网络多囊肾病创新管理中心主任York Pei教授分享了来自ADPKD遗传学研究的新见解。随后,他接受肾医线专访,进一步探讨了遗传学研究与遗传学检测在ADPKD诊疗中的重要价值。现整理York Pei教授报告和采访要点,与读者分享。
York Pei 教授
- 加拿大多伦多大学肾病科医学教授
- 多伦多综合医院研究所高级科学家
- 加拿大多伦多大学健康网络多囊肾病创新管理中心主任
- 加拿大皇家内科医师学会会员[FRCP(C)]
- 美国内科医师学院院士(FACP)
- 美国肾脏病学会会员(FASN)
- 主要研究领域:遗传性肾脏疾病(尤其是常染色体显性多囊肾病[ADPKD])的遗传、基因组和转化研究。他还在家族性IgA肾病、家族性肾病综合征和Alport综合征的遗传研究方面做出了重要贡献。他已发表超过200篇经同行评议的文章,与国内外研究人员进行了广泛的合作,并培养了众多遗传性肾脏疾病领域的临床和研究学者。
- 2016年,York Pei教授创立了多囊肾病创新管理中心,该中心为ADPKD患者提供先进的诊断和新型治疗方法。目前,该中心正在跟踪或共同管理超过500例患者,其中95%以上的患者至少参与了一项研究项目。
- York Pei教授于2019年荣获多囊肾病Lillian Jean Kaplan国际奖,并于2020年荣获加拿大肾脏基金会的卓越研究奖章。
报告概要:来自ADPKD遗传学研究的见解
常染色体显性多囊肾病(简称ADPKD)是最常见的遗传性肾脏病,约每1000个新生儿中就有1例ADPKD。在已明确遗传病因的病例中,PKD1和PKD2基因的突变分别占据了约65%~70%和20%~25%的比例[1]。PKD2引起的多囊肾病较PKD1所致多囊肾病进展慢,发生终末期肾病(ESRD)的时间约晚20年[1]。此外,还存在多种其他罕见的囊性肾脏或肝脏疾病基因,它们可能导致与ADPKD相似的症状。这些囊肿性病例有5%~8%会发展为终末期肾病(ESKD)[2]。目前,托伐普坦已在多个国家被批准用于治疗高风险进展为ESKD的ADPKD患者。
ADPKD的致病基因与遗传异质性
目前,已明确ADPKD主要由两个基因突变引起:PKD1基因和PKD2基因。PKD1基因位于16号染色体短臂(16p13.3)上,编码多囊蛋白1(PC1),是ADPKD的主要致病基因,占遗传确诊病例的约80%~85%。PKD2基因则位于4号染色体长臂(4q22-23)上,编码多囊蛋白2(PC2),占遗传确诊病例的约15%~20%。此外,少数ADPKD病例可能由其他罕见基因突变引起。
ADPKD的基因突变类型多种多样,包括点突变、插入、缺失、重排等。这些突变可能导致蛋白质的结构和功能发生改变,进而影响囊肿的形成和发展。例如,框内插入突变、错义突变、同义突变属于非蛋白截短突变,无义突变、经典剪切位点突变、移码突变(碱基缺失)属于蛋白截短突变。
不同突变类型对疾病进展风险的影响存在显著差异。一项针对来自220个无亲缘关系的ADPKD家系的780例患者开展的前瞻性研究表明,与携带PKD1截短突变的患者相比,携带PKD1框内插入/缺失突变、PKD1非截短突变或PKD2突变的患者身高矫正的肾脏总体积(HtTKV)更小、ESRD风险和死亡风险更低(P<0.001)[3]。
此外,ADPKD还表现出高度的遗传异质性,即在不同患者甚至同一家族的不同成员中,即使携带相同类型的基因突变,其临床表现和疾病进展速度也可能存在显著差异。研究发现,约12%(45/375)的家系中出现了患病情况不一致的亲属对[4],这使得在个体患者层面进行ADPKD的风险预测颇具挑战性。ADPKD的这种遗传复杂性可能与基因突变的类型、位置、修饰基因以及环境因素等多种因素有关,PKD1等位基因的相互作用、基因间相互作用也是造成ADPKD遗传复杂性的原因。
由PKD1和PKD2截短突变的双亲遗传引起的多囊肾病极为罕见。Monique Losekoot等确定并报道了6个疑似体细胞嵌合体的家系[5],这些家系的患者表现为新发疾病或不对称多囊肾病(PKD)。其中一个家系中,先证者为新发非对称性PKD,55岁时患者肾功能正常。其患病女儿在19岁时双侧肾脏增大。先证者的磁共振成像显示左侧多囊肾脏明显增大,右侧肾脏轻度增大。panel的Sanger测序结果显示,先证者的女儿存在PKD1基因1 bp移码缺失,先证者未见PKD1基因缺失。然而,PCR产物的定量分析表明,先证者女儿的突变等位基因和WT等位基因的拷贝数大致相等,但先证者(母亲)的突变等位基因仅为WT等位基因的约1/10,这与先证者是体细胞嵌合体的观点一致。
ADPKD的发生机制:二次打击模型
ADPKD中,肾脏的主要结构改变即囊肿的形成。那么,囊肿是如何形成的呢?ADPKD的囊肿形成具有局灶性特点,每个囊肿都是由单个上皮细胞的克隆扩增而成[6,7]。ADPKD囊肿形成与年龄相关,随着年龄增长囊肿不断增多增大。
ADPKD的发生机制尚未完全阐明。Stephen T. Reeders于1992年首次提出ADPKD中囊肿形成的“二次打击模型”。Y Pei的研究发现,肾小管中的所有上皮细胞都含有相同的生殖系PKD突变。在单个上皮细胞中发生了“野生型”PKD等位基因的体细胞突变,该上皮细胞的克隆性增殖导致肾小管扩张和囊肿形成,囊内液体积聚(图1)[8]。ADPKD在细胞水平上是隐性的,其原因是多囊蛋白信号传导减少。可能还存在其他导致PKD信号传导减少的过程。
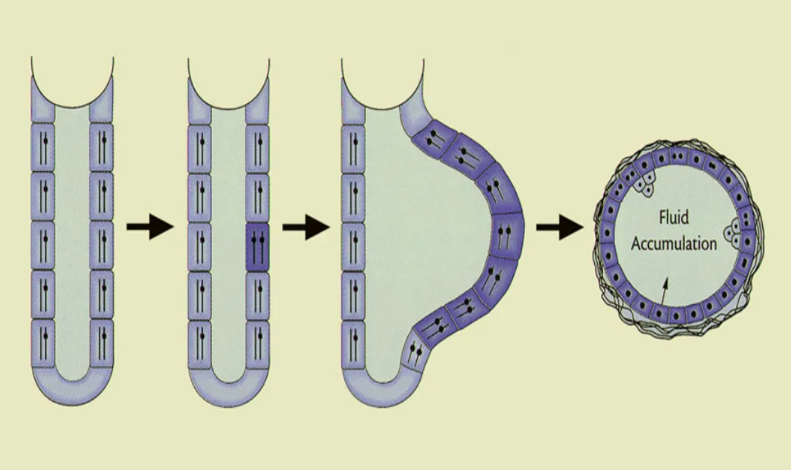
图1. ADPKD囊肿形成的"二次打击"模型
Zhang等通过对来自22例无亲缘关系ADPKD患者的90个囊肿进行全基因组测序检测,发现93%(84/90)的囊肿上皮细胞中存在PKD1/PKD2体细胞突变[9]。不同细胞基因剂量对PKD严重程度的影响不同,双基因及其相互作用、局部和随机因素也可能影响囊肿发生的阈值。此外,多囊蛋白为服务蛋白(Client Proteins)。通过对显性遗传性孤立性多囊肝病(PCLD)队列进行全外显子组测试,Whitney Besse等在三个基因ALG8、GANAB和SEC61B中发现了杂合子功能丧失突变,这些基因编码的蛋白质是在内质网中蛋白质生物合成途径不可或缺的蛋白质[10]。对体外细胞模型进行基因敲除分析显示,每个基因的失活都会导致PC1的成熟或转运缺陷,除这一共同特征外,每个基因对PC1的影响存在显著差异。SEC61B的缺失导致PC1的缺乏程度显著高于其他任何多囊性肝病相关基因(包括SEC63和PRKCSH)的缺失。ALG8的敲除导致PCI的低糖基化,而GANAB的敲除则导致N-聚糖部分的葡萄糖修剪缺陷,从而使PC1的表观分子量增加。
ADPKD的遗传学诊断
ADPKD的遗传学诊断主要依赖于基因检测。随着基因测序技术的不断进步,二代测序(NGS)技术的应用,使得ADPKD的遗传诊断更加快速、准确和全面。多伦多PKD扩展遗传流行病学研究(eTGESP)是一项针对单一地理区域(GTA,人口680万)的大规模研究,涉及1606 个家系的2171例患者[11]。研究发现75%的家系存在PKD1和PKD2突变,10%的家系中有非PKD1、非 PKD2囊性疾病基因(如 ALG8、ALG9等基因)突变,7.2%的家系存在杂合子遗传突变,15%的家系未检测到突变。在已确定遗传病例中PKD1和PKD2突变类型的比例分别为:PKD1蛋白质截短型(PT)占46%、PKD1插入缺失型(INDEL)占6%、PKD1非蛋白质截短型(NT)占 24%,PKD2占24%。这一研究为ADPKD的遗传学研究提供了人群数据支持。
初级纤毛在多囊肾病发病中的作用
根据临床和影像学评估,18%(32/174)的携带PKD1蛋白质截短突变患者病情较轻,提示PKD1截短突变可能存在保护性修饰效应[12]。
初级纤毛是一类微管介导的突起状细胞器,存在于大多数细胞表面,作为化学感受器和/或机械感受器发挥作用。初级纤毛在ADPKD病理生物学中处于核心地位,其初级纤毛的结构与功能异常可导致多囊肾病。初级纤毛是多囊蛋白复合体发挥功能的主要部位,目前已有超过20种多囊蛋白被发现在位于纤毛蛋白复合体[13]。此外,纤毛形成相关的基因突变也会导致多囊肾病(如Kif3a)。Chao Zhang等的研究发现,Glis2是多囊蛋白信号转导的早期效应因子,也是多囊肾病的治疗靶标[14]。
总之,对携带PT PKD1突变且病情轻微的患者进行WGS/WES,以识别纤毛通路中的罕见高影响突变,这可能为阐明保护性修饰效应提供一种有前景的方法。而对保护性修饰效应的遗传学见解可能改善临床预后,并为ADPKD的治疗提供新的靶点。
专家视点:遗传学研究在ADPKD诊断、治疗中担任重要角色
肾医线:ADPKD是ESKD最常见的单基因病因。遗传学研究在揭示ADPKD疾病机制中具有怎样的作用?
肾医线:遗传学研究在ADPKD的诊断和肾脏疾病严重程度方面扮演了什么角色?
然而,尽管这些突变类型能够反映出一种平均效应,且在具有相同突变类型的患者群体之间并不存在变异性,但疾病的严重程度却存在差异。这一现象揭示了修饰效应的存在。修饰效应可能具有双重性:一方面,它可能由于额外的遗传因素而加剧整体疾病的严重程度,表现出有害性;另一方面,它也可能发挥保护作用,即某些额外的遗传因素能够减轻疾病的严重程度。目前,我们虽然能够根据突变类型对患者进行大致的预后判断,但这种判断方法并非百分百准确。
肾医线:遗传学检测可以提供ADPKD的诊断和预后信息。哪些患者适合进行遗传学检测?
York Pei教授:目前,ADPKD的诊断主要依赖于肾脏影像学检查,因此通常无需进行遗传检测。然而,对于那些囊肿变化不典型的患者,遗传检测可能会提供重要信息,因为他们的病变模式可能并不完全符合常规的诊断标准,举例来说,当超声检查显示囊肿数量不足以确诊,或者当患者没有ADPKD家族史时,就可能出现这种情况。在没有ADPKD家族史的情况下,不能使用基于影像学的诊断,因为单纯依赖影像学检查进行诊断的准确性会大大降低。此外,在某些情况下,患者可能表现出一些与ADPKD典型特征不符的临床症状,这时遗传检测可能就显得尤为重要。
肾医线:遗传学检测如何支持ADPKD的治疗?
参考文献
[1] Clin J Am Soc Nephrol. 2021 May 8;16(5):790-799.[2] J Am Soc Nephrol 29: 13-23, 2018.[3] J Am Soc Nephrol 27:1861-68, 2016.[4] KI Rep 5: 1828-32, 2020[5] Kidney Int Rep (2020) 5, 1828–1832.[6] Cell 87: 979-87, 1996[7] J Clin Invest 99: 194-9, 1997.[8] Trends Mol Medicine 7: 151-6, 2001.[9] J Am Soc Nephrol. 2021 Dec 1;32(12):3114-3129.[10] Besse W et al JCI 127: 1772-85, 2017.[11] Journal of the American Society of Nephrology 33(11S):p 147-148, November 2022.[12] CJASN 16: 379,2021.[13] Watnick T and Germino G. Nature Genetics 34: 355-6, 2003.[14] Nat Commun. 2024 May 1;15(1):3698.
- 推荐文章